For generations, sickle cell disease has been one of the most difficult inherited blood disorders to manage. It affects millions of people around the world and has long been associated with recurring pain crises, frequent hospital visits, progressive organ damage, and a shorter life expectancy. Although advances in medicine have helped improve symptom management, many patients still spend their lives planning around the unpredictable nature of the disease. For families affected by sickle cell disease, the possibility of living without constant fear of the next pain episode has often felt out of reach.
That is why a recent milestone in Louisiana has attracted national attention. Twenty-three-year-old Daniel Cressy became the first person in the state to be declared functionally cured of sickle cell disease after receiving an FDA-approved CRISPR gene-editing therapy known as Casgevy. His recovery represents far more than an individual success story. It demonstrates how years of scientific research are beginning to translate into life-changing treatments for people born with inherited disorders. While this therapy is not suitable for every patient and still comes with significant challenges, it marks an important step toward changing what living with sickle cell disease could look like in the future.
A Lifetime Shaped by Sickle Cell Disease
Daniel Cressy was diagnosed with sickle cell disease as an infant after his parents noticed symptoms while he was still a toddler. The condition quickly became part of everyday life for his family. He frequently developed high fevers, severe pain throughout his chest and abdomen, and episodes that required emergency medical care. Hospital stays became a familiar experience rather than an occasional event.
Reflecting on those early years, Daniel shared, “I was diagnosed as an infant and my parents noticed symptoms when I was just a toddler when I would cry nonstop, without relief. So they would be forced to take me to the hospital and I would usually stay for at least a few nights at a time.” As he grew older, those hospitalizations continued. During childhood, he was admitted between six and twelve times each year because of complications from the disease.
Although his symptoms gradually became easier to manage during high school, sickle cell disease continued to influence his daily life. Physical activities that demanded endurance often left him exhausted, and he had to be mindful of situations that could trigger a pain crisis. Despite those obstacles, Daniel remained focused on building a future beyond his diagnosis instead of allowing the condition to define his ambitions.

One Dream Led Him to Gene Therapy
Daniel had always wanted to become a commercial airline pilot. Flying represented both a personal passion and a career that matched his determination to tackle difficult challenges. However, that dream appeared impossible because Federal Aviation Administration medical standards did not permit individuals with active sickle cell disease to obtain the certification required to fly commercially.
Rather than accepting defeat, Daniel contacted the Federal Aviation Administration to ask whether there was any path that would allow him to qualify in the future. The response gave him something he had never expected. “If I could cure my Sickle Cell Disease either through a bone marrow transplant or through gene therapy, then I could become a pilot.” Around the same time, gene-editing treatments were beginning to emerge as a promising option for patients with severe sickle cell disease.
When Manning Family Children’s Hospital established its gene therapy program, Daniel discussed the opportunity with his medical team. The treatment process required more than two years of preparation, insurance approvals, stem cell collection, laboratory modification of his cells, chemotherapy, and careful monitoring. Although lengthy, every stage brought him one step closer to pursuing the career he had imagined for years.
How Casgevy Changes Blood Cells
Casgevy is one of the first FDA-approved therapies to use CRISPR/Cas9 gene-editing technology for sickle cell disease. Instead of correcting the original mutation responsible for the disease, scientists modify a different genetic switch that controls the production of fetal hemoglobin, a type of hemoglobin naturally produced before birth.
People with sickle cell disease experience problems because their adult hemoglobin causes red blood cells to become rigid and crescent shaped. Fetal hemoglobin behaves differently. It helps red blood cells remain flexible and greatly reduces the sickling process responsible for painful blockages inside blood vessels. By increasing fetal hemoglobin production, many of the disease’s most serious complications can be prevented.
The treatment begins with collecting the patient’s own blood-forming stem cells. Those cells are sent to a specialized laboratory where CRISPR technology edits them before they are returned to the treatment center. After chemotherapy prepares the bone marrow to receive the modified cells, they are infused back into the patient’s bloodstream. Once established inside the bone marrow, the edited stem cells begin producing healthier red blood cells that contain much higher levels of fetal hemoglobin.
This approach differs from a traditional bone marrow transplant because patients receive their own genetically modified cells instead of donor cells. Using the patient’s own stem cells helps avoid complications such as graft-versus-host disease while reducing the need to find a compatible donor.

Why Doctors Call It a Functional Cure
Daniel’s treatment has been widely described as a functional cure, but that term has a very specific meaning in medicine. Unlike a complete genetic cure, the therapy does not erase the inherited mutation that causes sickle cell disease. Instead, it changes how Daniel’s blood-forming stem cells function, allowing them to produce large amounts of fetal hemoglobin. This healthy form of hemoglobin helps prevent red blood cells from becoming sickle shaped, greatly reducing or even eliminating the painful complications associated with the disease.
For Daniel, the results became evident within months of receiving the modified stem cells. After completing chemotherapy and the stem cell infusion, he spent several weeks recovering under close medical supervision before continuing outpatient follow-up appointments. Three months after the procedure, his doctors reported that his hemoglobin levels were the highest they had ever been. Speaking about his recovery, Daniel said, “I am great internally. I am physically fit as well. My hemoglobin is over 16. But externally I still am recovering. Like I am still bald.”
His remarkable improvement was celebrated during a ceremony at Manning Family Children’s Hospital on June 22, where he rang the ceremonial bell to mark the completion of his treatment. Surrounded by his family, physicians, hospital staff, and Louisiana leaders, Daniel described this new phase of his life as “Life 2.” He also reflected on what the experience has meant to him personally, saying, “I feel like God chose me to be the first one in the state because my story, once I do finally become a commercial pilot, is going to be inspirational for a lot of people. Overcoming what seemed impossible became my greatest blessing. While many spend their lives searching for purpose, mine found me. Now, instead of looking for meaning, I can spend my life fulfilling it.”
A Breakthrough That Still Comes With Challenges
Daniel’s story offers hope, but gene-editing therapy is not yet a treatment that every person living with sickle cell disease can receive. Casgevy involves an intensive medical process that includes stem cell collection, chemotherapy, hospitalization, and months of follow-up care. Patients must also meet strict medical criteria before they can be considered eligible for treatment, and the procedure is currently available only at specialized treatment centers.
Access remains another major challenge. Because Casgevy is a relatively new therapy with a very high price tag, obtaining insurance approval can be difficult and time-consuming. Daniel experienced those obstacles firsthand. Although he was ultimately approved for treatment, the process required months of discussions between his healthcare team and his insurance provider before authorization was granted.
Looking back on that experience, Daniel acknowledged how emotionally demanding the approval process became. “I wasn’t denied gene therapy. I just had to go through extra hoops because it was new and expensive. Trying to navigate approval took a lot out of me, but my friends who are flying jets and fighting sickle cell all kept me going. The team at Manning Family is amazing too.” His experience reflects a broader issue that many patients may face as healthcare systems work to make these advanced therapies more widely available.
Researchers also continue to study the long-term effects of gene editing. While clinical trial results have been encouraging, patients will continue to be monitored for many years to determine how durable the treatment remains and whether additional medical considerations emerge over time. As more people receive therapies like Casgevy, physicians will gain a clearer understanding of their long-term safety and effectiveness.
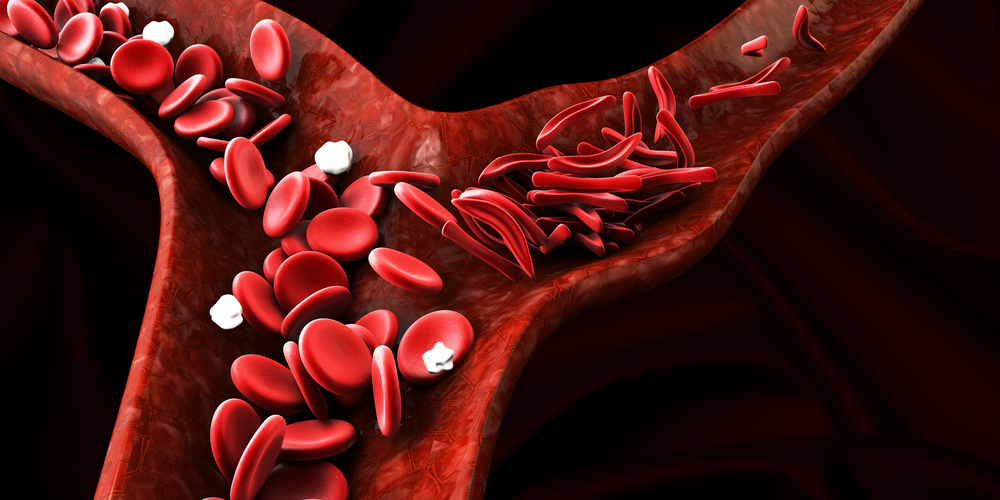
What This Means for the Future of Sickle Cell Disease
The approval of CRISPR-based therapies represents one of the most significant advances in sickle cell treatment since the disease was first described more than a century ago. Until recently, most therapies focused on reducing pain crises, preventing complications, or managing symptoms after they occurred. Gene editing offers a completely different approach by addressing the biological mechanism responsible for the disease itself.
That does not mean conventional care is becoming obsolete. Many people living with sickle cell disease will continue to depend on medications, blood transfusions, regular medical monitoring, healthy lifestyle habits, and comprehensive support from hematology specialists. Gene therapy is currently appropriate only for selected patients, and expanding access will require continued investment in specialized treatment centers, insurance coverage, and patient education.
Daniel hopes his journey encourages others to remain optimistic about the future. He has also established the Privileged Pilots Project, a nonprofit organization dedicated to helping aspiring pilots facing medical or life challenges pursue their goals while raising awareness about sickle cell disease. Encouraging eligible patients to explore emerging treatment options with their healthcare providers, he said, “I suggest they keep at it and be very persistent on making sure your care team understands how much the treatment means to you. This therapy is worth it because your second life, Life 2, will be amazing.”
More Than a Medical Milestone
Daniel Cressy’s recovery represents far more than an individual success story. It reflects decades of scientific research, collaboration, and innovation that have brought gene editing from the laboratory into real-world patient care. For many families affected by sickle cell disease, this milestone offers renewed optimism that future generations may experience fewer painful crises and greater opportunities to live full, independent lives.
At the same time, Daniel’s experience reminds us that groundbreaking treatments do not immediately erase every challenge. Expanding access, improving affordability, and ensuring that eligible patients can benefit from these advances remain important priorities. As researchers continue refining gene-editing technology, stories like Daniel’s provide a glimpse of what may become possible for many more people living with inherited blood disorders in the years ahead.




